.svg "\"\"")







 Navigating the complexities of ISO 13485:2016 can seem daunting, but understanding its requirements is crucial for medical device manufacturers aiming for Quality Management System (QMS) excellence. This blog explores the clauses of the standard and how best to implement a digital eQMS solution that can meet their demands.
Navigating the complexities of ISO 13485:2016 can seem daunting, but understanding its requirements is crucial for medical device manufacturers aiming for Quality Management System (QMS) excellence. This blog explores the clauses of the standard and how best to implement a digital eQMS solution that can meet their demands.
What is ISO 13485?
ISO 13485:2016 is the internationally recognised standard that specifies requirements for a medical device quality management system. It requires companies to develop documented systems that ensure the highest levels of safety, consistency and traceability from design and development to manufacturing and post-market surveillance.
What are the 8 clauses of ISO 13485?
ISO 13485:2106 comprises 8 clauses:
1. Scope.
2. Normative References.
3. Terms and Definitions.
4. Quality Management System.
5. Management responsibility.
6. Resource management.
7. Product realisation.
8. Measurement, analysis and improvement.
Of these, clauses 4 - 8 cover the major, mandatory requirements of the standard.
What are the 5 key clauses of ISO 13485?
Clause 4: Quality Management System (QMS)
At the core of ISO 13485 is the requirement to build a documented QMS (Quality Management System).
General requirements
This section specifies the essential mechanics of a QMS. It defines how it should support your business as it works in cycles of PDCA (Plan, Do, Check and Act) to drive a process of continual quality assurance.
It also introduces the concept of risk-based thinking that should inform the way you approach quality management in your organisation.
Documentation requirements
Clause 4 goes on to define how your QMS should control documents and records to create products that exactly match specifications and regulatory demands - while generating the required evidence of compliance for auditors and regulators.
- Documentation and records management: A robust system for creating, reviewing, and maintaining documents and records is essential. This includes standard operating procedures (SOPs), work instructions, and records that demonstrate compliance with regulatory requirements and the effectiveness of the QMS. Your QMS should facilitate easy access to current documents and secure archival of obsolete ones, ensuring full traceability and accountability.
- Control of documents: Procedures must be in place for document approval, review, and updates. The QMS should manage revisions and ensure only the current versions are accessible to relevant personnel.
- Control of records: Records must be identifiable, retrievable, and protected against loss, damage, and unauthorised access. Records demonstrate that you designed and manufactured your product exactly as specified.
This section also specifies the production of two key pieces of documentation:
Quality Manual
Your quality manual describes the scope of your QMS and the hierarchy of documentation in your system. The manual defines how all your QMS procedures should work together to generate the documents and records that can prove your products have been specified, designed and manufactured according to requirements and regulations. It demonstrates how quality cascades downwards through your system:
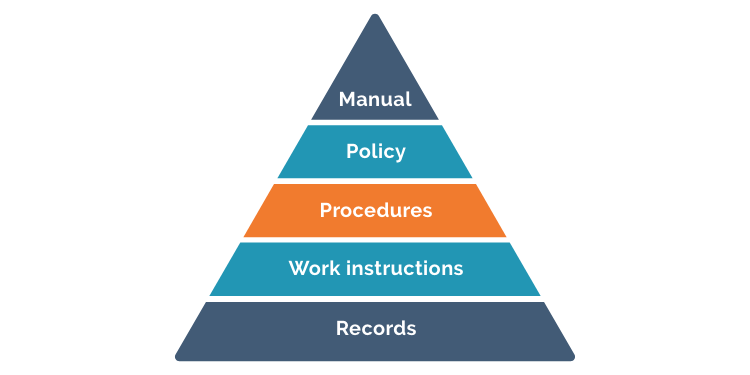
Medical Device File
The standard defines the content requirements for the medical device file (aka the Device Master Record in the FDA QSR). The file shall include:
- Description of the product, including intended use and indications for use.
- Product labelling and instructions for use.
- Specifications for the product.
- Specifications and procedures for manufacturing, inspection, labelling, packaging, storage, handling, and distribution.
- Specifications for measuring and monitoring.
- Specifications and procedures for product installation (if applicable).
- Procedures for product servicing (if applicable).
Download your free Medical Device File Creation Procedure Template
Clause 5: Management responsibility
Top management's role is critical in the effectiveness of the QMS:
- Commitment and leadership: Leadership must demonstrate a commitment to developing and implementing the QMS and continually improving its effectiveness. This includes communicating the importance of meeting regulatory and customer requirements throughout the organisation.
- Policy and objectives: Establishing a quality policy that is aligned with the organisation's purpose and the expectations of its customers. Quality objectives should be measurable and consistent with the quality policy.
- Roles, responsibilities, and authorities: Clearly defining and communicating the organisation's roles, responsibilities, and authorities to ensure effective QMS processes.
Clause 6: Resource management
The standard emphasises the need for adequate resources, which include:
- Personnel: Ensuring that all personnel involved in quality processes are competent based on education, training, skills, and experience. This might include conducting training programs and setting up digital tools for self-attestation to build data for compliance.
- Infrastructure and work environment: Providing the necessary infrastructure (facilities, equipment, software) and work environment to support product requirements. This includes managing the work environment to ensure product safety, particularly in clean rooms or controlled environments for certain medical devices.
Clause 7: Product realisation
This involves the entire process of bringing a medical device from concept to delivery:
- Planning: Establishing quality objectives and requirements for the product and planning the stages of product development.
- Design and Development: Applying systematic design and development processes, including risk management, verification, and validation activities to ensure the product meets specified requirements.
- Production and Service Provision: Implementing controlled conditions for production, including monitoring and control of equipment, facilities, and materials to ensure product conformity.
- Purchasing and Supplier Management: Assessing and selecting suppliers. Continuously evaluating supplier performance to ensure they meet the required quality standards and regulatory requirements. Implementing purchasing controls.
- Delivery: Ensuring that the final product is properly packaged, labelled, and delivered in a way that maintains its integrity and conformity.
Download our flexible Supplier Scorecard Template (easy-to-use Excel format )
Clause 8: Measurement, analysis, and improvement
Continuous improvement is a cornerstone of ISO 13485:
Monitoring and Measurement: Regularly monitoring and measuring critical aspects of the QMS and product to ensure conformity to product requirements and QMS effectiveness. This includes feedback mechanisms, internal audits, and monitoring of production and service processes.
Analysis of Data: Analysing data gathered from monitoring activities to identify trends, opportunities for improvement, and the need for corrective or preventive actions.
Improvement: Implementing actions to improve processes based on data analysis and outcomes of audits and reviews. This includes corrective actions to address nonconformities and preventive actions to eliminate potential non-conformities.
Why ISO 13485 matters
Quite apart from ensuring the quality and safety of your end products, gaining ISO 13485 is often a prerequisite for gaining regulatory approval. For example, you’ll likely need ISO 13485 to be granted a CE marking by a Notifying Body in the EU.
In the same way, the harmonisation of FDA 21 CFR Part 820 and ISO 13485 will soon make the standard the required stepping stone for every developer in the US, the world’s largest medical device market.
9 reasons you need to digitise and automate for ISO 13485 compliance
The modern medical device development process can be fraught with complexity. In the new era of IoT, implantables, SaMD (software as medical device) and generative AI, developers are generating huge amounts of design, testing and safety documentation within complex, multi-team sprints.
Companies need to digitise and automate their processes to manage all this documented information to meet the demands of ISO 13485 for control and traceability.
1. Enhanced document control
Automation ensures that all documents are easily accessible, up-to-date, and controlled according to ISO 13485 requirements. It eliminates the risks of lost or outdated documents, facilitating better management of document lifecycles.
2. Improved traceability
An automated QMS enhances the traceability of products throughout their lifecycle, a key requirement of ISO 13485. It enables accurate tracking of design changes, manufacturing processes, and distribution paths.
3. Consistent compliance
Automating compliance-related tasks, like CAPA (Corrective and Preventive Actions) and audits, ensures that these critical processes are conducted in a timely and effective manner, aligning with ISO 13485 standards.
4. Streamlined processes
Automation standardises processes across the organisation, reducing variability and ensuring consistent adherence to quality procedures, crucial for ISO 13485 compliance.
5. Increased efficiency
Automated workflows reduce manual tasks, freeing up valuable resources and time that can be redirected towards innovation and improvement, thus speeding up the certification process.
6. Enhanced quality management
With an automated QMS, monitoring and measuring quality metrics become more straightforward, allowing for real-time quality management and improvements aligned with ISO 13485 requirements.
7. Better risk management
Automation provides tools for more effective risk management, a cornerstone of ISO 13485. It enables a systematic approach to identifying, evaluating, and mitigating risks associated with medical device manufacturing.
8. Audit readiness
An automated QMS keeps all necessary documentation and records audit-ready, simplifying the audit process required for ISO 13485 certification and ensuring that any required information is easily retrievable.
9. Scalability
As your company grows, an automated QMS can quickly scale to accommodate new products, processes, or regulatory requirements, ensuring continuous compliance with ISO 13485 as your business evolves.
What’s standing in the way of your company gaining ISO 13485?
But most medical device developers will understand all this. If you are currently using a paper-based QMS or coping with a DIY digital approach, you will be acutely aware of how difficult it is to prepare such a system for auditing by a notified body.
Yet the complexity of many proprietary eQMS systems may put you off taking the leap.
The real challenge may be migrating to a closed-looped eQMS system - without creating huge amounts of work and bringing extra disruption to your operations.
You need the formal digital tools to meet the control and traceability requirements of ISO 13485 without changing the ‘way you do things’ just to meet the demands of a piece of QMS software.
In fact, auditors and regulators are keen on seeing Quality Management Systems that do not impose unnecessary processes because it inflates the risk of the system becoming too unwieldy to use effectively. The risk of ‘overprocessing’ can be as dangerous as a lack of control.
Take a lean approach to ISO 13485
The quest for a Lean eQMS should be a search for a system that integrates ISO 13485 compliance seamlessly into your business operations.
As you consider how to ready your business to gain ISO 13485, look for a partner and a set of digital tools that you can adopt and adapt to fit the way you work. You need a system that can act as a robust digital framework for compliance without taking months to implement or stopping your development in its tracks.

.jpg)

%20(1).webp?width=133&height=76&name=ISO%20IEC%2027001%20(1)%20(1).webp)